
Improving our detection of risk variants to disease
Published:
The Problem
Each Genome-wide association studies (GWAS) could provide insights into a biological mechanism that underlies the risk of disease in humans.(1) However, identifying these mechanisms has proved to be challenging. GWAS associations often include dozens of variants in linkage disequilibrium with one another that tag a single causal variant. Most causal variants do not directly alter protein-coding sequences but instead occur in noncoding gene regulatory elements such as enhancers, which can influence gene expression over long distances. (2,3,4,5) Furthermore, common diseases appear to involve contributions from multiple cell types, and many enhancers appear to act in specific cell types or states.
The Solution
To address this, our collaborators recently developed the activity-by-contact (ABC) model to predict which enhancers regulate which genes and generated genome-wide maps of enhancer-gene linking to interpret GWAS variants in 131 human cell types and tissues. Utilizing GWAS variants experimentally validated to link to inflammatory bowel disease (IBD), I built a pipeline (ABC-Max) to easily calculate how enriched causal variants are in predicted enhancers. Using this methodology, we found that causal variants are enriched in predicted enhancers by more than 20-fold in particular cell types such as dendritic cells (Fig1).
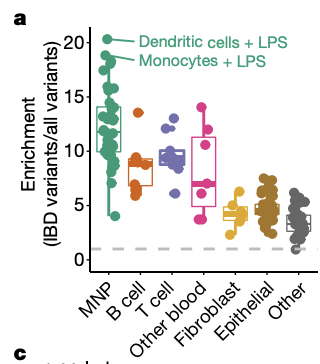
Fig 1. ABC maps connect fine-mapped variants to enhancers, genes and cell types. a, Enrichment of fine-mapped IBD variants (PIP ≥ 10%) in ABC enhancers (left) and all other accessible regions (right) in each of the 131 biosamples. MNP, mononuclear phagocytes. Box plots show the median (middle line) and interquartile range (boxes) and whiskers show observations less than or equal to quartiles ± 1.5× the interquartile range.
These variant-to-function maps reveal an enhancer that contains an IBD risk variant and that regulates the expression of PPIF to alter the membrane potential of mitochondria in macrophages (Fig2).
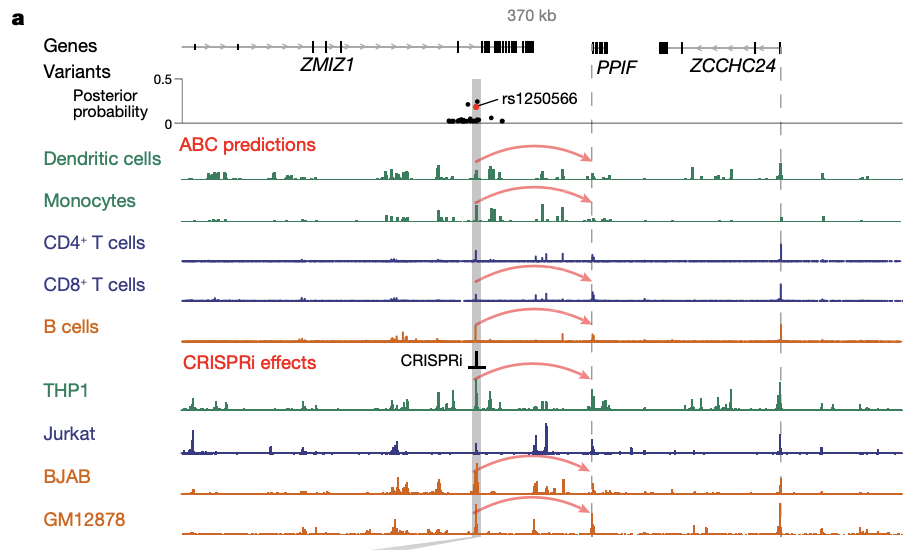
Fig 2. An IBD risk variant (rs1250566) overlaps with an enhancer that is predicted to regulate PPIF. Signal tracks represent chromatin accessibility (from ATAC-seq (assay for transposase-accessible chromatin using sequencing) or DNase-seq (DNase I hypersensitive site sequencing)). Grey bar, enhancer containing rs1250566; dashed lines, TSSs; red arrows for dendritic cells, monocytes, CD4+ and CD8+ T cells and B cells, ABC-Max predictions; red arrows for THP1, Jurkat, BJAB and GM12878 cells, CRISPRi leads to a significant decrease in PPIF expression.
Our study reveals principles of genome regulation, identifies genes that affect IBD and provides a resource and generalizable strategy to connect risk variants of common diseases to their molecular and cellular functions.
Data
We utilized GWAS data in (7) to validate these enhancer-gene links in IBD.
References
1.Claussnitzer, M. et al. A brief history of human disease genetics. Nature 577, 179–189 (2020). 2.Farh, K. K.-H. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015). 3.Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). 4.Gasperini, M., Tome, J. M. & Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292–310 (2020).
- van Arensbergen, J., van Steensel, B. & Bussemaker, H. J. In search of the determinants of enhancer–promoter interaction specificity. Trends Cell Biol. 24, 695–702 (2014). 6.Fulco, C.P., Nasser, J., Jones, T.R. et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat Genet 51, 1664–1669 (2019).
- Huang, H. et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547, 173–178 (2017).